
 7月4日,国家药品监管局和国家卫生健康委联合发布《定制式医疗器械监督管理规定(试行)》(以下简称试行规定),以满足临床实践中的罕见特殊个性化需求,规范定制式医疗器械监督管理,保障定制式医疗器械的安全性、有效性。试行规定共分为总则、备案管理、设计加工、使用管理、监督管理、附则六章三十五条,于2020年1月1日起正式施行。
所谓定制式医疗器械,是指为满足指定患者的罕见特殊病损情况,在我国已上市产品难以满足临床需求的情况下,由医疗器械生产企业基于医疗机构特殊临床需求而设计和生产,用于指定患者的、预期能提高诊疗效果的个性化医疗器械。
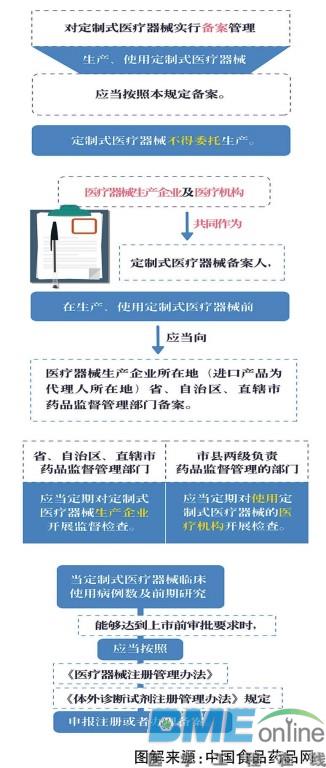
由于定制式医疗器械仅用于特定患者,数量极少、没有足够的人群样本进行临床评价,难以通过现行注册管理模式进行注册。考虑到产品特点,试行规定明确,对定制式医疗器械实行备案管理。根据试行规定,医疗器械生产企业及医疗机构共同作为定制式医疗器械备案人,在生产、使用定制式医疗器械前应当向医疗器械生产企业所在地(进口产品为代理人所在地)省(区、市)药品监管部门备案。省(区、市)药品监管部门应当定期对定制式医疗器械生产企业开展监督检查。市、县两级负责药品监管的部门应当定期对使用定制式医疗器械的医疗机构开展检查。
试行规定同时要求,当定制式医疗器械临床使用病例数及前期研究能够达到上市前审批要求时,应当按照《医疗器械注册管理办法》《体外诊断试剂注册管理办法》规定,申报注册或者办理备案。符合伦理准则且真实、准确、完整、可溯源的临床使用数据,可以作为临床评价资料用于注册申报。
为合理控制定制式医疗器械风险,试行规定对生产、使用定制式医疗器械的生产企业和医疗机构均提出了明确要求,并要求定制式医疗器械不得委托生产。根据试行规定,生产企业应当有定制式医疗器械研制、生产所需的专业技术人员,具备定制式医疗器械研制能力和研究基础,有相同类型的依据标准规格批量生产的医疗器械注册证及相应生产许可证,有相同类型的依据标准规格批量生产的医疗器械的生产能力和生产经验,并符合相应的质量管理体系。使用定制式医疗器械的医疗机构则要求为三级综合或者三级专科医院,具有与使用的定制式医疗器械相适应的诊疗
项目;有在医疗机构注册的、能够使用定制式医疗器械的主诊医师;具备使用同类已上市产品的经验,已开展同种疾病研究和治疗,临床专业水平国内先进;具备较高的医疗器械管理水平,已建立完善的医疗器械使用质量管理体系,具备医疗器械使用评价和医疗器械不良事件监测能力等。
试行规定还明确了定制式医疗器械设计生产及说明书标签相关要求,并且规定在保护患者隐私情况下,生产企业应当将定制式医疗器械产品设计环节延伸到医疗机构。在要求定制式医疗器械研制、生产符合医疗器械生产质量管理规范及相关附录要求外,试行规定还对人员、设计开发、质量控制、追溯管理等方面提出特殊要求。医疗机构应当对使用后的定制式医疗器械开展评价工作,制定完善的安全防范措施和风险控制计划。定制式医疗器械不得在大众传播媒介进行广告宣传,除法律法规允许外,禁止将患者信息用于生产和使用定制式医疗器械以外的其他用途等。
根据试行规定,定制式医疗器械不包含患者匹配医疗器械。患者匹配医疗器械是指医疗器械生产企业在依据标准规格批量生产医疗器械产品基础上,基于临床需求,按照验证确认的工艺设计和制造的、用于指定患者的个性化医疗器械(例如定制式义齿),应当按照《医疗器械注册管理办法》《体外诊断试剂注册管理办法》的规定进行注册或者备案,注册/备案的产品规格型号为所有可能生产的尺寸范围。
据悉,目前,我国已有多家临床机构与医疗器械生产企业、高等院校合作开展了定制式医疗器械在骨科、颅颌面外科、整形外科、口腔修复科和眼科等领域的研发应用。
(来源:中国医药报)
|







 NB300、350、380维修手册
NB300、350、380维修手册 2025年职能部门月考核细则(医学工程部)
2025年职能部门月考核细则(医学工程部)





 浙公网安备 33010402001000号
浙公网安备 33010402001000号